


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
松本 淳
no journal, ,
We have developed a computational approach to build an atomic model from an electron microscopy (EM) image of a biological molecule. In this approach, many atomic models of the molecule with different conformations are prepared first by deforming the X-ray crystal structure or the modeled structure using a computational technique. Then, a variety of orientations is given to each atomic model to obtain projection images. Finally, the projection images are compared to the EM image. The atomic models with the projection similar to the EM image are regarded as the candidates for the atomic structure of the molecule. In the symposium, the application to the giant cadherin proteins, which are involved in the cell adhesion, will be explained.
石田 恒
no journal, ,
To understand the transition path between different reaction states and the free-energy profile along the path, a hybrid-simulation using all-atom molecular dynamics (MD) simulations and electron microscopy (EM) density maps was developed. The hybrid-simulation comprises two stages; one is to predict a transition path starting from an X-ray structure to EM density maps using an all-atom MD simulation in water medium. The other is to sample conformations to obtain the free-energy profile along the predicted path. The hybrid-simulation was applied to the system of ribosome-tRNAs-EFG to understand the mechanism of tRNA translocation. The results showed that a ratche-like motion, the movement of the P/E-gate and EF-G play important roles in tRNA translocation.
米谷 佳晃
no journal, ,
Precise structural information about biomolecular hydration has become available with recent high-resolution X-ray and neutron experiments. Although further information, kinetics of hydration water, is also important to understand the relevant dynamical processes, it is not yet fully clarified. One of the long-standing unsolved problems is the distinct timescale feature of hydration water. Some cases show a hydrogen-bond lifetime of a few pico-second, but in other cases it is over ~100 ps. What causes such a lifetime difference? Here, we carried out molecular dynamics simulations for various systems, DNA, protein, and ions in water, to explore the origin of the lifetime difference. Based on our results, we will also discuss our prospect of a future pKa study.
村上 洋
no journal, ,
逆ミセルは、無極性溶媒中で界面活性剤分子の自己組織化により形成され、ナノメートルスケールの球殻の中に水を含む。逆ミセルのサイズは実験的に制御可能であり、蛋白質分子やDNAを含む、水溶性分子をその中に溶かすことが可能である。そのため、逆ミセルを用いて、水や導入分子の物性のナノ空間拘束効果がこれまで広範に調べられてきた。最近我々は、界面活性剤AOTと溶媒イソオクタンを用いて調製した半径1nm程度の逆ミセルにおいて、導入色素分子の周りの水の拡散運動が室温付近で凍結しており、また、水媒質の格子緩和エネルギーが逆ミセル中の水分子数の少なさに起因して小さいことを示した。そこで、我々は逆ミセル中色素分子のホールバーニング分光を着想した。室温で色素分子の周りがガラス的であれば、永続的ホールバーニングが可能であり、また、小さな格子緩和エネルギーは、ホールバーニングスペクトル幅の先鋭化に導くと考えられる。ホールスペクトルの逆ミセルサイズやホールバーニングのための照射レーザー波長依存性の結果と、色素溶液や色素高分子膜の結果も示し議論する。
松尾 龍人; 荒田 敏昭*; 小田 俊郎*; 藤原 悟
no journal, ,
F-アクチン及びミオシンS1タンパク質とその水和水のピコ秒領域のダイナミクスを中性子準弾性散乱によりJ-PARCを用いて解析した。その結果、F-アクチン構成原子はミオシン構成原子よりも高頻度で揺らいでいることが分かった。また、水和水のスペクトル解析から、F-アクチン水和水の運動性がミオシン水和水よりも大きいことが明らかとなった。これらの結果は、F-アクチンとその水和水の協同的作用がF-アクチンの高い運動性を作り出し、ミオシン結合に最適な構造を素早く探索できることを示唆している。
角南 智子; 河野 秀俊
no journal, ,
Conformational flexibility of DNA plays important roles on biological processes such as transcriptional regulations. Therefore, it is important to analyze when and how DNA shows conformational variations. Electron density maps in crystallographic analyses contain information of conformational variations. But such information is typically discarded in the refinement process. To extract the information about conformational variations, we have performed comprehensive analysis of the electron density maps of DNA crystals. We found that positive FoFc densities are observed more frequently around phosphates than around bases, indicating high flexibility of DNA phosphates. In the presentation, we will discuss more details.
池部 仁善; 桜庭 俊*; 河野 秀俊
no journal, ,
In eukaryotic cells, genome DNA is stored in a complex with histone proteins (H3, H4, H2A and H2B). Acetylation to terminal regions of histones (histone tails) is generally believed to regulate gene expression through dissociation of tails from DNA, although conformations of disordered tails remain poorly understood. In this work, we examined differences in conformational ensembles of H3 tail with or without K14 acetylation using adaptive lambda square dynamics simulation. The result suggested that the acetylation does not make the tail dissociate from DNA. Instead, it enhanced secondary structure formation of the tail and unwrapping of DNA from the structured histone core regions. This study elucidated the first step of the gene regulation mechanism.
藤原 悟; 松尾 龍人; 山田 武*; 柴田 薫
no journal, ,
In order to investigate the regulatory mechanism of muscle contraction in terms of protein dynamics, we carried out neutron scattering experiments on the native thin filaments (NTF) in the presence and absence of Ca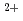 and F-actin using the dynamics analysis spectrometer
and F-actin using the dynamics analysis spectrometer  at J-PARC. The elastic incoherent and quasielastic neutron scattering experiments showed that NTF in the -Ca state is more flexible than in the +Ca state, and that this difference in flexibility arises from the different distributions of the local atomic motions. Comparison with F-actin suggests that the differences arise from the regulatory proteins. These results imply that regulation of the protein dynamics plays an important role in the regulatory mechanism of muscle contraction.
at J-PARC. The elastic incoherent and quasielastic neutron scattering experiments showed that NTF in the -Ca state is more flexible than in the +Ca state, and that this difference in flexibility arises from the different distributions of the local atomic motions. Comparison with F-actin suggests that the differences arise from the regulatory proteins. These results imply that regulation of the protein dynamics plays an important role in the regulatory mechanism of muscle contraction.